CASE REPORT |
https://doi.org/10.5005/jp-journals-10030-1363 |
Patient with Severe Traumatic Brain Injury and Malaria in a Middle Eastern Country
1-3Department of Trauma Surgery, Hamad General Hospital, Doha, Qatar
Corresponding Author: Corresponding Author: Vishwajit Verma, Department of Trauma Surgery, Hamad General Hospital, Doha, Qatar, Phone: +447771742108, e-mail: vverma@hamad.qa
ABSTRACT
Aim and objective: This manuscript reports a case of severe traumatic brain injury in a patient complicated by Falciparum malaria with review of the literature and discussion of possible underlying mechanism aggravating the secondary brain injury.
Background: Plasmodium falciparum (P. falciparum) malaria accounts for more than 90% of deaths caused by malaria in the world. Parasitized red blood cells sequester and cause capillary occlusion, endothelial damage, cytokine activation, and dysregulation of coagulation leading to exacerbation of secondary injury after traumatic brain injury.
Case description: The case reports highlight a rare case of 38-year-old patient admitted after traumatic brain injury with rapid deterioration. Patient was found to have P. falciparum malaria with 3.9% parasitised RBC. CT head revealed a large right sided subdural haemato-hygroma, midline shift and ischemia in the right posterior cerebral artery territory. Despite timely intervention and evacuation of subdural haematoma, patient had a poor outcome with multiple infarcts, hemorrhagic transformation, and early hydrocephalus. Patient remains in a vegetative state in a long-term unit.
Conclusion: Combined effect of endothelial, microcirculatory, inflammatory, and clotting dysfunction caused by traumatic brain injury and parasitaemia leading to aggravation of secondary brain injury.
Clinical significance: This case report highlights the intricate relationship between endothelial, inflammatory, and coagulation cascade triggered after brain injury. The nature of coagulopathy in such patients is complex with state of hypo-coagulation early followed by a hyper-coagulatory state in the late phase.
How to cite this article: Verma V, Alansari ANH, Arumugam S. Patient with Severe Traumatic Brain Injury and Malaria in a Middle Eastern Country. Panam J Trauma Crit Care Emerg Surg 2021;10(3):134-138.
Source of support: Nil
Conflict of interest: None
Keywords: Falciparum, Ischemia, Malaria, Pathophysiological mechanism, Traumatic brain injury, Traumatic subdural hematoma.
BACKGROUND
Annual incidence of traumatic brain injury is high in Middle Eastern countries. Hamad General Hospital (HGH) is the only level 1 trauma center in the state of Qatar, admitting more than 300 patients with severe traumatic brain injury every year.1 Qatar’s has a large population of expatriate workers representing myriad nationalities and ethnic groups. Malaria is still a leading cause of morbidity and mortality in the developing world. World Health Organization's world malaria report 2020, stated that more than 400,000 people die of malaria with Plasmodium falciparum (P.falciparum) accounting for more than 90% of the world’s malaria mortality.2 P. falciparum is transmitted by female Anopheles mosquitoes. The parasite once transmitted undergoes development inside the liver. The hepatic phase is followed by the erythrocyte cycle that is responsible for the clinical manifestation of the disease. Malarial infection can lead to brain damage. It has been postulated that cerebral manifestations of malaria are underpinned by occlusion of brain capillaries due to swelling of endothelium and sequestration of infected red blood cells (IRBCs).3,4 Endothelial injury leads to activation of clotting cascading resulting in ischemia and microhemorrhages. Here we describe a patient who presented with severe traumatic brain injury and P.falciparum malaria.5
While TBI and malaria are common in many parts of the world, we could not find a single report of their combination. We report a rare case of a patient admitted with severe TBI complicated by malaria, review the literature and discuss the possible underlying mechanisms.
CASE DESCRIPTION
Patient is a 38-year-old male who recently travelled from Uganda to Qatar with history of fever, headache, and abdominal pain. Patient was feeling dizzy and fell down while trying to walk up to the toilet. His initial Glasgow Coma Score (GCS) reported to be 15/15 with bilaterally reactive pupils. Patient’s GCS dropped rapidly during the transport to trauma resuscitation unit. On arrival in trauma resuscitation unit patient developed anisocoria with a GCS of 3/15 and was intubated with rapid sequence intubation. An urgent trauma whole body CT scan revealed a large subdural haemato-hygroma causing mid-line shift of 16 mm and multiple frontal and temporal contusions with effacement of basal cisterns (Figs 1A and B). Admission CT also showed a subtle hypodensity in the posterior cerebral artery territory (Fig. 1C). Patient was taken for a lifesaving evacuation of subdural hematoma. In view of the history of travel, fever and low platelets on presentation, malaria screen was also performed in addition to routine blood tests. Patient was transferred to trauma intensive care unit after the evacuation with anisocoric pupils that were sluggishly reacting to light. Patient’s conventional clotting tests on arrival showed low platelet count of 43,000 per microliter. Remaining conventional clotting tests were within normal limit. Rotation thromboelastometry showed mild increase in clot formation time (CFT) of 149 sec with mild reduction in amplitude 10 minutes after clotting time (A10) of 43 seconds.
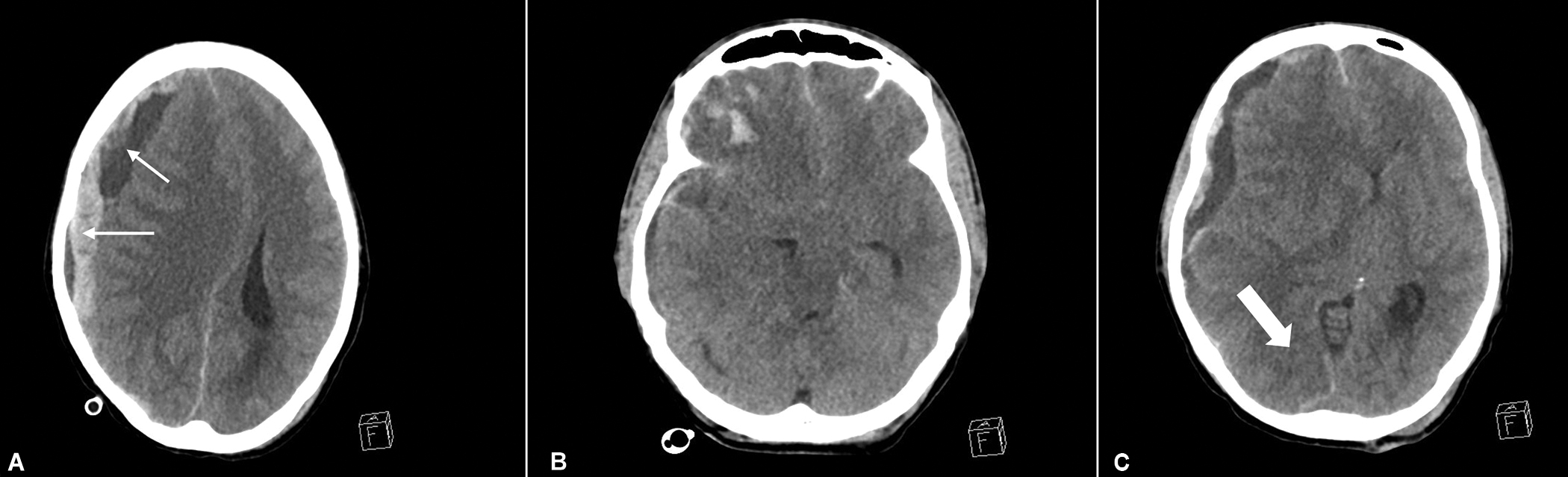
Figs 1A to C: (A) Axial sections from first CT brain at the time of admission showing a large right-sided subdural hematohygroma (Thin white arrows), midline shift and frontal contusions; (B) Section at the level of lower midbrain and upper pons, showing bilateral uncal herniation and obliterated perimesencephalic cisterns; (C) Subtle hypodensity is seen in the posterior cerebral artery territory (Thick white arrow) and diencephalic shift
Patient received multiple transfusion of platelets both in intraoperative and postoperative period to maintain platelets above 80,000 per microliter. The malaria screen confirmed presence of P. falciparum in early trophozoite ring stage with 3.9% parasitized red blood cells. Antimalarial treatment was commenced on antimalarial treatment in the form of Atovaquone and Proguanil. Patient underwent a repeat CT brain on day 2 of the admission that revealed evolving infarcts in the posterior cerebral artery territory (Figs 2A to D) with interval evolution of temporal contusions on the right side (Fig. 2C).

Figs 2A to D: (A) Post right-sided subdural evacuation CT scan; (B) Established posterior cerebral artery territory infarction; (C and D) Infarction seen in vein of Labbe territory (white arrow)
Patient remained in a state of eyed close coma and GCS of 3/15 with intact bilateral corneal, vestibulo-ocular, cough reflexes, and spontaneous respiration. Deep venous thrombosis chemoprophylaxis was commenced after 24 hours. On 4th day of the admission, patient underwent per-cutaneous tracheostomy. A repeat CT head was performed on the 5th day of admission due to bilateral increment in size of his pupils. The CT scan revealed a nonobstructive hydrocephalus, established infarcts in MCA, and PCA territory with a new intraparenchymal bleed in the posterior temporal lobe. exerting a mass effect (Fig. 3). External ventricular drain (EVD) was inserted to treat hydrocephalus. CT head was done before EVD was removed on 11th July 2021 that showed resolution of hydrocephalus, established infarcts in anterior, and posterior vascular territory including midbrain and regression of intraparenchymal hemorrhage. He was discharged from ICU after removal of the EVD and remains in a stable, self-ventilating condition with no neurological recovery showing extensor posturing, mid-sized unreactive pupils, and an eye closed coma in a long-term facility (Fig. 4).
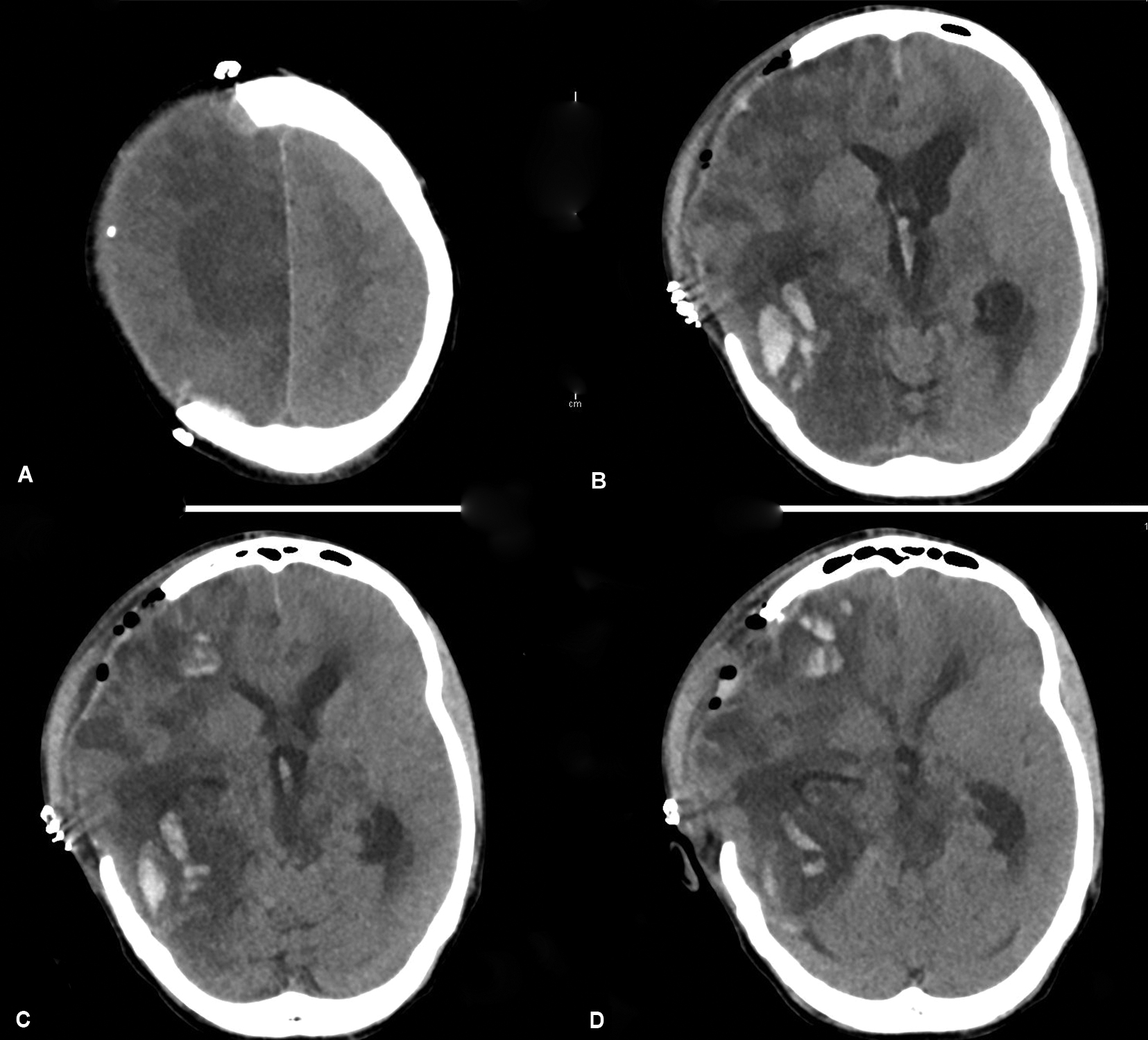
Figs 3A to D: Hemorrhagic conversion of the infarcted area; (A) Hypodensity in the anterior cerebral artery territory; (B to D) Hemorrhagic conversion of the infarcted area
DISCUSSION
This case above describes a rare case of traumatic brain injury in a patient with P. falciparum malaria. We hypothesize that this patient’s outcome was adversely affected by the presence of falciparum malaria. Cerebral malaria is defined by the World Health Organization as a clinical syndrome characterised by a coma which persists longer than 1 hour after termination of a seizure or correction of hypoglycemia with the presence of asexual forms of parasites on peripheral blood smears and no other cause to explain the coma.6 The patient described above did not meet the criteria for cerebral malaria before the injury however the presence of malarial infection may have exacerbated the secondary brain injury.
The pathophysiology of neuronal injury caused by cerebral malaria is not well understood. Much of the insight into the pathogenesis comes from autopsy studies,7 which probably represents the most severe manifestation of the illness. Sequestration of IRBCs in the cerebral microvasculature is believed to be the key factor in pathogenesis of brain injury in cerebral malaria. The process of sequestration starts with the adherence of IRBCs to the intercellular adhesion molecule-1 (ICAM-1) in the vascular endothelium facilitated by P.falciparum erythrocyte membrane protein-1 (PfEMP) expressed on the surface of infected erythrocytes.3 The sequestration is self-perpetuating due to the adherence of noninfected RBCs with the infected RBCs leading to the formation of rosettes. The process of sequestration and formation of rosettes causes perfusion deficit with reduction in the supply of the neural substrates (glucose and oxygen) leading to neuronal death responsible for both acute and chronic sequelae of the cerebral malaria. Furthermore, the parasitic antigen released at schizogony stage triggers a cytokine and chemokine response, which potentiate neuronal injury.4 P. falciparum malaria infection is also accompanied by a state of dysregulation of coagulation characterized by low platelets, low circulating levels of anti-coagulants, generation of activated thrombin, and pro-coagulant microparticles.5 Clinical manifestation of the complex syndrome as spontaneous major intracranial hemorrhage albeit rare has been reported. The pattern of bleeding reported ranges from SAH, subdural to intracerebral hemorrhage and even extradural hemorrhage.8,9,10,11,12,13 A prospective study14 of 21 patients with P. falciparum malaria correlated imaging characteristics with postmortem findings, found seven patients with normal CT scan, eight with diffuse cerebral edema of which two died. Both patients had renal failure and one had disseminated intravascular coagulation. Five patients out of 21 had cerebral edema with bilateral thalamic and cerebellar hypoattenuation on CT brain with no survivors. These patients had a median day one APACHE II score or 26 and failure of two or more organs. Postmortem examination of these five patients showed diffuse cerebral edema with multiple petechial hemorrhages scattered diffusely in the brain.
Histopathological examination revealed perivascular aggregates of parasitized red blood, inflammatory cells, and necrotic tissue with ring hemorrhages and parasitised red blood cells in cerebral blood vessels.
Activation of host immune system in response to an infection results also in activation of components of hemostatic system of an organism. The presence of P. falciparum infected erythrocytes (PfiE) triggers the upregulation of adhesion molecules on the endothelial surface increased production of tumor necrosis factor (TNF) which is a potent trigger of tissue factor initiating extrinsic clotting pathway.4,15 Thrombocytopenia is a consistent feature of acute P. falciparum infection which results from a combination of activation, removal by the reticuloendothelial system and consumption of platelets in different steps of coagulation.16 Patchy endothelial damage seen in these patients is theorized to be caused by apoptosis due to adhesion of PfiE on TNF-stimulated endothelial cells.19
Moreover, traumatic brain injury results in a state of coagulation dysregulation with both hypo-coagulopathy and a prothrombotic tendency occurring simultaneously.20 Nearly two-thirds of the patient with severe TBI, show clotting abnormalities on conventional tests of clotting.21 Impact on the brain results in shearing and rupture of the microvessels resulting in contusions. Peri-contusional area where the effect of injury is lower triggers mechanosensitive molecular pathway thereby starting a delayed disruption of microvasculature and blood brain barrier.22 Damage to microvessels and blood brain barrier results in activation and subsequent consumption of platelets leading to thrombocytopenia. The other characteristic of brain injury is the hyper-fibrinolysis, thought to be caused by exaggerated activation of clotting cascade via tissue factor (TF) with local release of endogenous tissue-type plasminogen activator (tPA) and urokinase-type plasminogen activator (uPA) from the contusional brain injury.23 Increase in fibrinolysis is indicated by increase in concentrations of fibrinogen degradation products and predicts progression of intracranial hemorrhage.24
Our patient presented with a severe traumatic brain injury with rapid neurological deterioration. Patient underwent decompressive craniectomy in a timely fashion and despite that he suffered a poor outcome. Low platelets, history of fever, and travel prompted us to investigate him for malaria. Postdecompression patient developed infarction inside multiple vascular territories on the side of injuries. Patient had subsequent hemorrhagic transformation of the infarction. Patient’s outcome was worsened by combined effects of malarial parasite on the brain with coagulation dysregulation caused by both malaria and traumatic brain injury.
The mechanical shift of the brain and herniation of medial temporal lobe across the tentorium cerebri causes the compression of posterior cerebral artery against the rigid tentorium. The incidence of PCA territory infarcts varies from 9-88%. Middle cerebral artery infarction is caused by gross herniation or cerebral edema or direct vascular injury. Gross mass effects can produce stretching and compression of fine lenticulostriate and thalamo-perforators resulting in infarctions.25 The patient described above developed ischemia very early, even the first CT head showed early ischemic changes in the PCA territory.
Patient’s poor neurological outcome and a state of eye closed unresponsiveness with intact vegetative functions is most probably explained by compression of PCA and resultant ischemia of the midbrain.
Patient later developed a new intraparenchymal hemorrhage in inferior right temporal lobe. As this bleed was located in the drainage area of inferior mesenteric vein also known as vein of Labbe (Fig. 4A)26 it is possible that is this was a hemorrhagic conversion of a venous infarct (Figs 4B and C )27 The vein of Labbe injury is caused by direct impact or counterblow to the cranium and is associated with temporal bone fracture. One case series reported that vein of Labbe injury was associated with temporal bone fracture in 15 out of 16 patients.28 Vein of Labbe can lead to traumatic cerebral infarction and poor outcome.28 Venous infarcts can also be caused by compression of the vein due to compression by the mass lesion. It is also possible that an exaggerated coagulation dysregulation due to malaria and traumatic brain injury resulted in the venous thrombosis and subsequent hemorrhagic conversion.
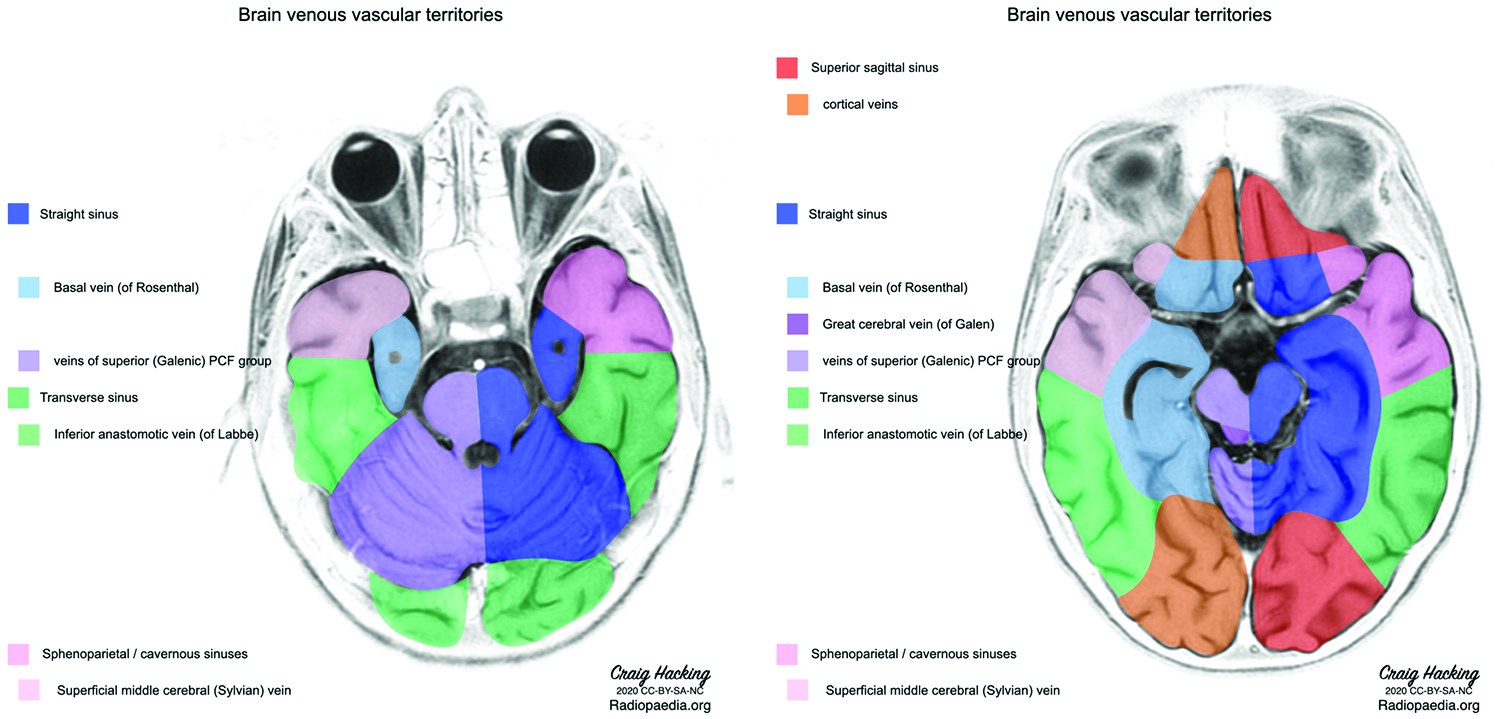
Fig. 4: Brain venous vascular territories showing the drainage area of vein of Labbe or inferior anastomotic vein. (Image reproduced under creative common license, Case courtesy of Assoc Prof Craig Hacking, Radiopaedia.org, rID: 80,107)
CONCLUSION
This manuscript repots a rare case of a patient with P. falciparum malaria, that complicating a traumatic brain injury. Our literature search did not reveal any other case reports of traumatic brain injury complicated by falciparum malaria.
The presence of malarial parasite in blood and its effect on the brain worsened the secondary brain injury. Patient demonstrated coagulopathy manifested by thrombocytopenia at the time of admission to the hospital. The thrombocytopenia persisted well into the postoperative period despite multiple blood and products transfusion. Patient developed multiple, anterior, posterior cerebral artery, and possible venous infarcts were possibly due to a combination of mass effect caused by subdural haematoma and clotting dysregulation due malaria and severe traumatic brain injury.
This case provides a valuable insight into the intricate and complex relationship that exists between inflammation and clotting pathways both triggered after injury and probably amplifies by presence of malarial infection. The case also highlights the clinical complexity underpinned by dysregulated homeostasis and accelerated rate of secondary injury and challenges faced by the treating physicians.
Orcid
Vishwajit Verma https://orcid.org/0000-0003-2428-2391
REFERENCES
1. El-Menyar AConsunji RAl-Thani H. Handbook of Healthcare in the Arab World. Published online 2021:2469-2482. DOI: 10.1007/978-3-030-36811-1_106
2. World Malaria Report. https://www.who.int/publications/i/item/9789240015791
3. Newton CRJCHien TTWhite N. Cerebral malaria. J Neurology Neurosurg Psychiatry 2000;69(04):433-441. DOI: 10.1136/jnnp.69.4.433
4. Idro RJenkins NENewton CR. Pathogenesis, clinical features, and neurological outcome of cerebral malaria. Lancet Neurol 2005;4(12):827-840. DOI: 10.1016/s1474-4422(05)70247-7
5. Moxon CAHeyderman RSWassmer SC. Dysregulation of coagulation in cerebral malaria. Mol Biochem Parasitol 2009;166(02):99-108. DOI: 10.1016/j.molbiopara.2009.03.006
6. Severe falciparum malaria. World Health Organization T Roy Soc Trop Med H Communicable Diseases Cluster. 2000;94Suppl 1:S1-90.
7. Afandi DSampurna BSutanto I,et al. Autopsy findings in severe wmalaria - a case report. Medical J Indonesia 2008;17(03):210-215. DOI: 10.13181/mji.v17i3.315
8. Karanth SS, Marupudi KC, Gupta A. Intracerebral bleed, right haemiparesis and seizures: an atypical presentation of vivax malaria. Bmj Case Rep 2014;2014:bcr2014204833. DOI: 10.1136/bcr-2014-204833
9. SenthilKumaran SBalamurugan NSuresh P et al. Extradural hematoma in plasmodium vivax malaria: are we alert to detect? J Neurosci Rural Pract 2013;4(Suppl 1):S145-S146. DOI: 10.4103/0976-3147.116476
10. Dwarakanath SSuri AMahapatra AK. Spontaneous subdural empyema in falciparum malaria: a case study. J Vector Dis 2004;41(3-4):80-82.
11. Murugavel KSaravanapavananthan SAnpalahan A et al. Subarachnoid haemorrhage in plasmodium falciparum malaria. Postgrad Med J 1989;65(762):236-237.
12. Gall CSpuler AFraunberger P. Subarachnoid hemorrhage in a patient with cerebral malaria. New Engl J Med 1999;341(08):611-613. DOI: 10.1056/nejm199908193410814
13. Seshadri PDev AVViggeswarpu S et al. Acute pancreatitis and subdural haematoma in a patient with severe falciparum malaria: case report and review of literature. Malaria J 2008;7(01):97-97. DOI: 10.1186/1475-2875-7-97
14. Patankar TFKarnad DRShetty PG et al. Adult cerebral malaria: prognostic importance of imaging findings and correlation with postmortem findings. Radiology 2002;224(03):811-816. DOI: 10.1148/radiol.2243010588
15. Hunt NHGrau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol 2003;24(09):491-499. DOI: 10.1016/s1471-4906(03)00229-1
16. Kelton JGKeystone JMoore J,et al. Immune-mediated thrombocytopenia of malaria. J Clin Invest 1983;71(04):832-836. DOI: 10.1172/jci110836
17. Pongponratn ERiganti MHarinasuta T et al. Electron microscopy of the human brain in cerebral malaria. Southeast Asian J Tropical Medicine Public Health 1985;16(02):219-227.
18. Grau GEMackenzie CDCarr RA,et al. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J Infect Dis 2003;187(03):461-466. DOI: 10.1086/367960
19. Pino PVouldoukis IKolb JP,et al. Plasmodium falciparum-infected erythrocyte adhesion induces caspase activation and apoptosis in human endothelial cells. J Infect Dis 2003;187(08):1283-1290. DOI: 10.1086/373992
20. Laroche MKutcher MEHuang MC et al. Coagulopathy after traumatic brain injury. Neurosurgery 2012;70(06):1334-1345. DOI: 10.1227/neu.0b013e31824d179b
21. CRASH-3 trial Collaborators. Effects of tranexamic acid on death, disability, vascular occlusive events and other morbidities in patients with acute traumatic brain injury (CRASH-3): a randomised, placebo-controlled trial. Lancet 2019;394(10210):1713-1723. DOI: 10.1016/s0140-6736(19)32233-0
22. Monson KLConverse MIManley GT. Cerebral blood vessel damage in traumatic brain injury. Clin Biomech (Bristol, Avon) 2019;64:98-113. DOI: 10.1016/j.clinbiomech.2018.02.011
23. Kushimoto SShibata YYamamoto Y. Implications of fibrinogenolysis in patients with closed head injury. J Neurotraum 2003;20(04):357-363. DOI: 10.1089/089771503765172318
24. Zhang JHe MSong YXu J. Prognostic role of D-dimer level upon admission in patients with traumatic brain injury. Medicine (Baltimore) 2018;97(31):e11774. DOI: 10.1097/md.0000000000011774
25. Server ADullerud RHaakonsen M et al. Post-traumatic cerebral infaction. Neuroimaging findings, etiology and outcome. Acta Radiol 2001;42(03):254-260.
26. Ikushima IKorogi YKitajima M et al. Evaluation of drainage patterns of the major anastomotic veins on the lateral surface of the cerebrum using three-dimensional contrast-enhanced MP-RAGE sequence. Eur J Radiol 2006;58(01):96-101. DOI: 10.1016/j.ejrad.2005.11.011
27. Mageid RDing YFu P. Vein of Labbe thrombosis, a near-miss. Brain Circ 2018;4(04):188-190. DOI: 10.4103/bc.bc_34_18
28. Munakomi SKumar B. Case report: traumatic vein of Labbé hemorrhagic infarction - a distinct neurosurgical entity. F1000research 2015;4:586. DOI: 10.12688/f1000research.6910.1
________________________
© The Author(s). 2021 Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by-nc/4.0/), which permits unrestricted use, distribution, and non-commercial reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.